
Mucopolysaccharidose
Les mucopolysaccharidoses sont un type de trouble du stockage lysosomal dans lequel les molécules de sucre complexes ne sont pas dégradées normalement et s’accumulent en quantité dangereuse dans les tissus de l’organisme. Elles provoquent ainsi un faciès typique et des anomalies au niveau des os, des yeux, du foie et de la rate accompagnées parfois d’un retard mental. Des mucopolysaccharidoses se développent lorsque les parents transmettent à leurs enfants les gènes défectueux à l’origine de ces troubles.
Les mucopolysaccharidoses surviennent lorsque les enzymes nécessaires à la dégradation et au stockage des molécules de sucre complexes font défaut à l’organisme.
Habituellement, les symptômes comprennent une petite taille, l’hirsutisme, la rigidité des articulations des doigts et un aspect grossier du visage.
Le diagnostic est basé sur les symptômes, un examen clinique et des analyses de sang.
Le traitement dépend du type de trouble. Il peut inclure un traitement enzymatique substitutif à vie et, parfois, une greffe de moelle osseuse.
Bien que l’espérance de vie soit normale, certains types entraînent un décès prématuré.
Il existe différents types de troubles héréditaires. Dans de nombreux troubles métaboliques héréditaires, les deux parents de l’enfant affecté sont porteurs d’une copie du gène anormal. En général, comme deux copies du gène anormal (gène récessif) sont nécessaires pour que le trouble se développe, aucun des parents n’est généralement atteint de la maladie. Certains troubles métaboliques héréditaires sont liés à l’X, ce qui signifie qu’une seule copie du gène anormal peut causer la maladie chez les garçons.
Les molécules du glucose complexes appelées mucopolysaccharides (glycosaminoglycanes) sont essentielles à beaucoup de tissus de l’organisme. Dans les mucopolysaccharidoses, l’organisme présente un déficit des enzymes nécessaires à la dégradation (métabolisme) et au stockage des mucopolysaccharides. Il en résulte un excès de mucopolysaccharides dans le sang, excès qui se dépose dans des localisations inhabituelles de l’organisme.
Symptômes des mucopolysaccharidoses
Différentes formes de mucopolysaccharidose ont des symptômes légèrement différents, mais, en général, pendant la petite enfance et l’enfance, on peut remarquer une petite taille, l’apparition de poils épais et un développement anormal. Le visage peut sembler épais avec une grosse tête, un front proéminent, un nez court, des lèvres épaisses et une grosse langue. Les artères et les valves cardiaques sont parfois affectées. Les articulations des orteils sont souvent rigides,
Certaines formes de mucopolysaccharidose entraînent un retard mental qui peut s’aggraver au cours de la vie de la personne. D’autres types peuvent être caractérisés par une baisse de la vision ou de l’audition.
Transmission génétique
La maladie de Hunter est transmise par la femme selon un mode récessif lié à l'X, de ce fait seul les garçons sont atteints.
Le gène IDS est situé sur la partie télomérique du bras long du chromosome X dans la région dite Xq28.
A chaque grossesse les femmes conductrices ont un risque de 50 % d'avoir un enfant atteint s'il s'agit d'un garçon. L'étude de la généalogie permet de prévenir les femmes conductrices de cette maladie et de surveiller leur grossesse.
Tableau clinique
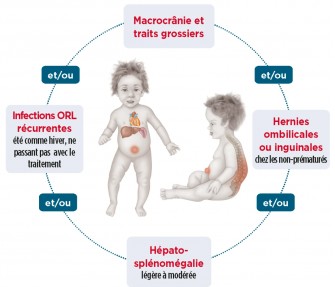
L'enfant est normal à la naissance, les signes n'apparaissant que progressivement. Le diagnostic est en général évoqué entre 2 et 4 ans, face à un garçon présentant notamment :
- encombrement rhinopharyngé permanent entrainant des infections répétées des voies aériennes supérieures
- faciés de Gargouille ou dysmorphie faciale
- cassure de la courbe pondérale
- abdomen hypotonique (hernies multiples)
Les autres symptômes :
- limitations articulaires (les mains en griffe)
- dysostose multiple
- nanisme
- hépatosplénomégalie
- atteinte cardiaque
- surdité
- atteinte respiratoire
- régression psychomotrice aboutissant à un retard mental. Il existe des formes modérées avec survie prolongée et intelligence conservée.
Le diagnostic biologique repose sur la mise en évidence de l'excrétion urinaire accrue de DS et HS et du déficit enzymatique (sérum, leucocytes, fibroblastes, trophoblaste ou amniocytes).
Traitement
- Chirurgie pour drainer l'excès de liquide céphalo-rachidien du cerveau
- Chirurgie pour libérer les nerfs et les racines nerveuses comprimés par des anomalies squelettiques et autres
- Chirurgie pour corriger les hernies
- Greffes de cornée pour améliorer la vision chez les patients présentant une opacification cornéenne importante
- Chirurgie pour enlever les amygdales et végétations adénoïdes pour améliorer la respiration chez les patients troubles obstructifs des voies respiratoires et OSA
- Thérapie de remplacement enzymatique (MPS I, II, IVA, VI et VII) pour réduire les symptômes non neurologiques et la douleur